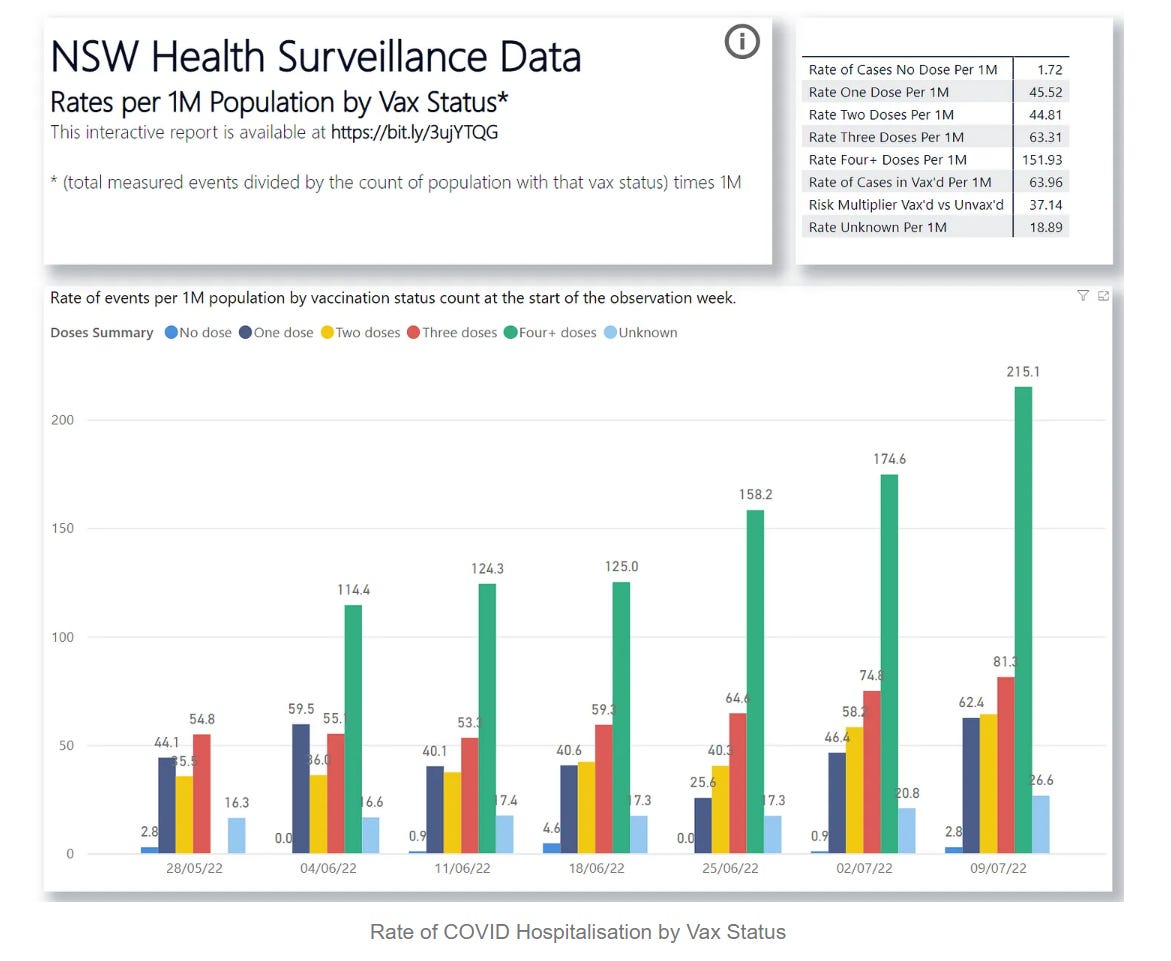
@Joes Place Update (Jan 2022): I have calculated a rigorous arrival curve of actual in-clade mutation rate for SARS-CoV-2 at 1.45 x 10-4 mutations per site per year. This analysis is taken straight from the Nextstrain data published 26 Jan 2022,35 and can be accessed by clicking here. The data extract and worksheet can be accessed by clicking here. This matches the GISAID estimate of rate of mutation (1.8 x 10-4 in Exhibit 7.8 below) well. Thus our assumption of 3.6 x 10-4 was conservatively suitable for this analysis. One thing to note as well, is that SARS-CoV-2 is not mutating nearly the rate at which SARS-CoV-1 did, nor is it mutating at the purported furious pace which was sold to us in the media (see bottom chart in Exhibit 7.8).
 Exhibit 7.8 – Actual mutation rate of SARS-CoV-2 compared to other human viruses
Exhibit 7.8 – Actual mutation rate of SARS-CoV-2 compared to other human viruses
There are 29,811 single strand RNA nucleotides in SARS-CoV-2. Given its 93 differential mutations from a ~March 2020 Alpha Clade 20B variant, which can be seen in Exhibit 7.7, this would at first glance indicate around 8.7 years of genetic distance wound up inside Omicron’s divergence from other existing variants. However we must adjust the calculation in that only 66 of the 93 total mutations constitute the most typical RNA virus mutation, called a ‘substitution’.36 Therefore,
 Exhibit 7.8 – Actual mutation rate of SARS-CoV-2 compared to other human viruses
Exhibit 7.8 – Actual mutation rate of SARS-CoV-2 compared to other human virusesThere are 29,811 single strand RNA nucleotides in SARS-CoV-2. Given its 93 differential mutations from a ~March 2020 Alpha Clade 20B variant, which can be seen in Exhibit 7.7, this would at first glance indicate around 8.7 years of genetic distance wound up inside Omicron’s divergence from other existing variants. However we must adjust the calculation in that only 66 of the 93 total mutations constitute the most typical RNA virus mutation, called a ‘substitution’.36 Therefore,
However, genetic distance by typical RNA virus mutation is not the end-all of this derivation. Not all nucleotides mutate at the same rate.37 As we just mentioned, most mutations arrive in the form of synonymous high frequency events called substitutions – mutations that separate Covid in-clade variants in linear sequence from their initial index sequence (see horizontal lines in the left panel of Exhibit 7.9 below). The mutations in Omicron do not follow this pattern, and in fact constitute an exception within the entire diagram. Instead, Omicron mutations comprise about 32% of what are called ‘insertion/deletion’ mutations (INDELs).38 INDELs do not arrive at the same rate as higher frequency substitutions, but rather constitute less common absences or novel-presences of an entire nucleotide. Insertions and deletions constitute a change in the logical structure of the RNA sentence and not a mere synonymous replacement of a word, if you will. Thus they produce failures (extinction) more often than successes, and as such constitute a much bigger challenge in terms of genetic language. They can also often result in different mutational clock measures as compared to those based upon substitutions alone. In fact, RNA virus INDELs are 4x less frequent in their occurrence than are substitutions (actually deletions are 8x less frequent, and the majority of INDELs for Omicron are deletions – however, we still use 4x here for conservancy).39 If we approach our genetic clock with this second method of measuring genetic distance, we get the following result – which importantly, substantiates and exceeds fairly well our substitution-nucleotide method of measuring genetic distance conducted above.Substitution Clock – .00036 x 29811 = 10.7 mutations per year 66/10.7 = 6.1 years of mutation
We therefore through triple-conservancy in this method,40 can reasonably cite that 6.1 years (the smaller of the two above substitution and INDEL based estimates) would be the minimum time duration required to enact all 66 Omicron substitution mutations as observed in sample sequence QLD-2568 of 2 Dec 2021. We must leave the alarming INDEL mutation rate in Wittgenstein silence because sadly we cannot connect it back to any kind of usable reference. Finally, before we move on from this set of calculations, we should employ this same method to take quick note of the evolutionary time which would be required for SARS-CoV-2 to have evolved naturally from its nearest relative among beta coronaviruses, BANAL52. This will act as a double-check of the validity of our estimates above as well (consilience). We divide by two here because two virus evolutionary pathways are involved in this analytical context. Reader please note that this is a benchmark comparison for establishing credibility of our assumed 3.6 x 10-4 survival mutation rate only. It does not mandate that SARS-CoV-2 necessarily evolved naturally from BANAL52. The conjectured Jan 2018 lab accident could have released either a natural or edited SARS-CoV-2 under our hypothesis.INDEL Clock – (.00036 / 4) x 29811 = 2.68 INDEL mutations per year 30/2.68 = 11.2 years of mutation
Total amount of time needed to naturally evolve from BANAL52 = (1/2 x 4% of 29,811 / 10.7) = ~55 years.